|
||||
|
|
ЛЕКЦИЯ № 12. Теоретическая электрохимия 1. Ассоциации в растворах электролитов. Понятие о теории сильных электролитов. Активность При увеличении концентрации растворов электролитов создаются условия, когда за счет электростатического взаимодействия становится возможным образование новых соединений – ассоциатов. На вероятность образования ассоциатов указал Н. Бьеррум, им определено то минимальное расстояние между ионами, при котором становится возможным образование ассоциатов. В симметричных электролитах образуются нейтральные ионные пары. Эти ионные пары имеют определенное время жизни, т. е. можно говорить об ионном равновесии между ионной парой и теми ионами, которые составляют эти ионные пары. Различают несколько видов ионных пар, находящихся в растворе: 1) константная K+, An–; 2) гидратно (сольватно)-ионная пара K+(H2O)An–; 3) гидратно (сольватно)-разделенная пара K+(H2O)N An–; 4) ионные тройники K+An–K+, An–K+An–. Образование ионных пар в растворе сказывается на величине проводимости раствора, причем появление и образование нейтральных ассоциатов снижает проводимость растворов электролитов. Для сильных электролитов, когда степень ионизации велика, константа ионизации зависит от концентрации, так как при накоплении в растворе большого числа ионов сказывается их взаимное влияние. Когда растворитель обладает малой диэлектрической проницаемостью (величина, показывающая, во сколько раз ослабевают силы взаимодействия электрических зарядов при расположении этих зарядов в диэлектрике по сравнению с силами в вакууме), создаются условия для электростатического взаимодействия сольватированных ионов противоположного знака. При этом последние подходят друг к другу на близкое расстояние и образуют ионную пару – сложный агрегат, состоящий из двух противоположно заряженных ионов, окруженных молекулами растворителя, в котором электрические заряды взаимно компенсированы, это – ассоциация. Параметр Бьеррума 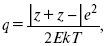 где е – заряд электрона –16 x 10–19Кл; к – константа Больцмана = 1.38 x 10–23Кдж/к; Т – абсолютная температура; Е – диэлектрическая проницаемость. Ассоциация ионов – образование из противоположно заряженных ионов (в растворах электролитов) особого рода частиц-ассоциатов, в которых ионы удерживаются за счет электростатического взаимодействия их электрических зарядов в соответствии с законом Кулона. Процессы ассоциации ионов подчиняются закону действующих масс, термодинамическим соотношениям. Простейшие ассоциаты – ионные пары, нейтральные или электрически заряженные, термодинамически устойчивые частицы, находящиеся в равновесии с простыми ионами.  Степень ассоциации ионов зависит от природы растворителя, электролита, температуры, концетрации раствора. Она возрастает с уменьшением диэлектрической проницаемости растворителя вследствие увеличения кулоновского притяжения ионов. Ассоциация – объединение однородных частиц в растворе. Сольватация – взаимодействие растворенного вещества с растворителем. Молекулярные группы, сольваты, образовавшиеся в результате взаимодействия, слой молекул растворителя, связанный с центральной частицей сольвата силами притяжения – сольватная оболочка. Наименьшее число молекул, удерживаемых в непосредственной близости от сольватированной частицы – координационное число сольватации. В зависимости от природы растворенного вещества, сольваты могут образовываться различными путями. Ион-дипольное взаимодействие – при растворении веществ с ионной структурой молекулы растворителя удерживаются около иона силами электростатического притяжения. Процесс гидратации – взаимодействие веществ с водой. 2. Термодинамика растворов электролитов. Типы ДЭС На границе раздела фаз электрод – раствор происходит перераспределение зарядов, в результате которого у поверхности электрода накапливается избыток ионов какого-либо одного знака, в результате на поверхности возникает избыток зарядов противоположного знака. Такое пространство разделения избыточных зарядов позволяет ввести понятие ДЭС. Условно можно представить две воображаемые плоскости, которые проходят через центры тяжести избыточных зарядов в поверхностном слое на электроде и раствора у поверхности электрода. Расстояние между плоскостями равно диаметру сольватированного комплекса (d). Таким образом, ДЭС можно рассматривать как плоский конденсатор: 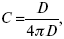 D – диэлектрическая проницаемость, для Н2О = 80, d = 10–8 – расстояние между обкладками. Перераспределяемый заряд на границе раздела фаз имеет динамическое преобладание. Согласно закону термодинамики, перенос вещества из одной фазы в другую характеризуется химическим потенциалом ? = ?Gхим ? зависит от концентрации частиц. ? = ?0 +RTlnС, где С – концентрация частиц, участвующих в реакции переноса; ?0– стандартное значение потенциала при С= 1. Согласно термодинамике, перенос вещества из одной фазы в другую происходит таким образом, что химический потенциал вещества действует до тех пор, пока ?1 = ?2 в обеих фазах. 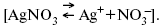 Определяющимися ионами являются ионы Ag+, следовательно, если химический ? ионов Ag (Ме), 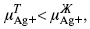 то, согласно уравнению Ле-Шателье, перенос ионов Ag+, в сторону раствора будет происходить с большей скоростью, чем в обратном направлении, в результате количество ионов Ag у поверхности будет увеличиваться, так как концентрация в растворе растет. ?Ag+ = ?0 + RT lnCAg+. Перенос происходит до тех пор, пока Когда наступит равенство ?, скорость переноса в обоих направлениях через границу раздела фаз становится одинаковой. Наступает равновесие; так как в переносе принимают участие заряженные частицы, то в этом случае говорят об электрохимическом равновесии. При установлении равновесия поверхность, с которой ушли Ag+, приобретает избыток отрицательного заряда, под действием его избыток положительных зарядов собирается в растворе у поверхности. Возникновение избытка заряда приводит к возникновению скачка ?на границе раздела фаз. Согласно представлению о плоском конденсаторе, скачок потенциала изменяется линейно с расстоянием от поверхности электрода. Емкость такого плоского конденсатора, когда ионная обкладка ДЭС состоит из катионов, составляет С = 20 МКФ/см2. 3. Современные подходы к описанию термодинамических свойств растворов электролитов При теоретическом подходе к концентрированным растворам электролитов предпринимались попытки уточнить классическую модель теории Дебая – Хюккеля за счет учета следующих эффектов: 1) собственного объема ионов; 2) изменения диэлектрической проницаемости вблизи ионов вследствие диэлектрического насыщения растворителя; 3) изменения макроскопической диэлектрической проницаемости в объеме раствора в зависимости от концентрации и т. д. Общий недостаток работ этого направления состоит в том, что в каждой из них учитывают только один или два из перечисленных эффектов. Расхождение современных статических теорий наблюдается, в основном, в ходе функций распределения на малых расстояниях. Теоретический расчет потенциала взаимодействия частиц на малых расстояниях сложен и не может быть пока проведен однозначно, так как на таких расстояниях, наряду с кулоновскими силами, играют роль квантово-механические, дисперсионные и другие силы. Г. Кеселером было развито специфическое взаимодействие ионов в растворах и показано, что некулоновские эффекты при сближении ионов проявляются вследствие десольватации ионов (при перекрытии сфер сольватации) и сольватации ионных пар как целого. Термодинамические характеристики ион-молекулярных взаимодействий в растворах Теория Дебая и Хюккеля позволила предсказать эффект выделения тепла при разбавлении растворов электролитов, вызванный тем, что при разбавлении уменьшается взаимодействие между ионами. Характеристики Химический потенциал 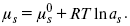 Средняя активность 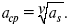 Коэффициент активности характеризует работу перенесения иона из идеального газа в реальный. Абсолютная активность 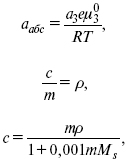 где МS– молярная масса растворенного вещества. 4. Термодинамические характеристики ионов в растворах электролитов ?i– химический потенциал иона. 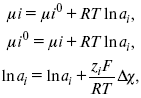 где ??изменение поверхностного потенциала данного раствора по сравнению с поверхностным потенциалом стандартного раствора; аi– «реальная» активность иона. 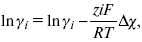 где ln?i– экспериментально определенный реальный коэффициент активного иона. ai = ?i mi. Поверхностная активность – способность вещества при абсорбции на границе раздела фаз понижать поверхностное натяжение. Адсорбция Г вещества и вызванное ею понижение поверхностного натяжения ?связаны с концентрацией с вещества в фазе, из которой вещество абсорбируется на межфазовую поверхность, – уравнение Гиббса. 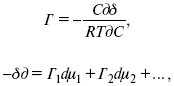 где д?/ дC– мера способности вещества понижать поверхностное натяжение на данной межфазной границе, называется также поверхностной активностью. 5. Неравновесные явления в ионной системе Равновесие в растворах электролитов всегда является динамическим, усредненным по времени, объему. Частицы раствора совершают хаотические движения, которые осуществляются периодическими перескоками с одного места на другое. В среднем эти перемещения частиц скомпенсированы так, что направленного макроскопического перехода ионов и диполей в условиях равновесия не происходит. Вызвать в растворе электролита неравновесные явления можно: 1) изменением активности растворенного вещества в одной части раствора по отношению к другой без изменения химического состава раствора; 2) наложением электрического поля, которое вызовет направленное перемещение заряженных частиц; 3) изменением состава раствора по отношению к равновесной концентрации возможных для данной системы веществ. В первом способе количество перескоков данной частицы iв направлении уменьшения ее концентрации оказывается больше, чем количество перескоков этой частицы в обратном направлении. В результате этого возникает поток диффузии. Во втором способе наложение электрического поля изменяет вероятность перескоков заряженных частиц по полю и против поля: у катионов количество перескоков по полю превышает количество перескоков против поля, у анионов – наоборот. Таким образом, в результате наложения электрического поля возникают потоки миграции катионов и анионов. Миграция ионов, составляющая основу электропроводности электролитов, сопровождает работу электрохимических систем. Общий поток ионов j = jд + jм, jд – поток диффузии, jм– поток миграции. Диффузионный потенциал – разность потенциалов, возникающая на границе двух растворов разных электролитов или двух растворов одного электролита, но разной концентрации. Диффузия в твердых электролитах характеризуется тем, что с повышением температуры скорость диффузии возрастает. Диффузия связана с механизмом образования вакансий и дислоцированных атомов. В результате колебаний около положения равновесия атомов, ионов, расположенных в узлах кристаллической решетки, некоторые из них, обладающие в данный момент избыточной энергией, могут покинуть свое положение равновесия и переместиться в соседние междоузлия. Ранее занимаемые ими места в решетке окажутся вакантными, кроме того, образуется соответствующее число дислоцированных атомов, такой процесс по Френкелю следует рассматривать как внутреннее испарение атомов твердого тела. Число вакансий, т. е. пустых мест в решетке, вообще может и не соответствовать числу дислоцированных атомов (атомов, расположенных в междоузлиях), так как вакансии могут образовываться на поверхности твердого тела в результате полного или неполного «испарения» поверхностных атомов. Каждой данной температуре соответствует определенное равновесное число вакансий, тем больше, чем выше температура. Наличие пустых мест в кристаллической решетке реального кристалла обеспечивает возможность процесса диффузии в твердых телах. Зависимость коэффициента диффузии от температуры: D = D0(–EaRT), где и – энергия активации процесса диффузии. Диффузия в полупроводниках. С понижением температуры проводимость убывает, при высоких температурах проводимость приближается к проводимости металла. Концентрация носителей тока (электронов проводимости) в металле практически не зависит от температуры, а в полупроводниках носители тока сами возникают в результате теплового движения. В полупроводниках, благодаря сильной связи валентных электронов с ядрами, положение атомов такое, что нужно сообщить энергию ионизации. Такая энергия поставляется тепловыми колебаниями атомов решетки. Число атомов с энергией, равной или превышающей энергию ионизации, мало. 6. Равновесие в системе жидкость – жидкость Равновесие жидкость – жидкость В случае ограниченной взаимной растворимости компонентов в жидком состоянии в двухкомпонентной системе осуществляется равновесие двух жидких фаз. Явление образования двух жидких фаз называется расслаиванием или расслоением. Типы диаграмм состояния систем с расслаиванием приведены в литературе по физико-химическому анализу. Взаимная растворяемость жидкостей при постоянном давлении зависит только от температуры. Рассмотрим случай возрастания растворимости с ростом температуры. При температуре больше критической обе жидкости смешиваются в любых соотношениях. Критические явления часто сопровождаются в момент перехода из двухфазного состояния в гомогенное образованием голубой опалесценции. Согласно правилу В. Ф. Алексеева (1886 г.), при разделении соединительных линий фазовой диаграммы пополам точки деления ложатся на общую прямую, которая заканчивается в критической точке растворения (прямая еК). Это правило позволяет дополнить экспериментальные данные о взаимной растворимости жидкостей, а также найти состав в критической точке по известным значениям tкр. Правило Алексеева приближенно, наиболее точно оно при выражении состава в процентах. Рассмотрим подробно наиболее распространенный тип систем с верхней критической температурой растворимости (ВКТР). Взаимную растворимость таких двухфазных систем изучают обычно визуально-политермическим методом, предложенным В. Ф. Алексеевым. Суть метода заключается в определении температуры перехода из расслоения в гомогенное состояние и обратно. Заготавливают ряд смесей определенного состава, помещают их по очереди в термостат (термостойкий стакан с водой или глицерином, снабженный термометром) и наблюдают изменение фазовых состояний системы с изменением температуры. При непрерывном встряхивании отмечают температуру, при которой мутноватая смесь становится прозрачной. Затем, охлаждая жидкость, отмечают температуру, при которой она вновь становится мутной (появление первых капелек второй фазы). Для удобства наблюдения за происходящими в смеси изменениями пользуются лампой-осветителем. Повторяют опыт с той же смесью несколько раз, причем добиваются, чтобы температуры появления и исчезновения мути были достаточно близки (расхождения не превышали 0,5о). Среднее значение между ними считают за температуру перехода. Этот простой метод позволяет по полученным экспериментальным данным построить бинодальную кривую и, следовательно, определить составы равновесных фаз. 7. Понятие ДЭС. Модельные представления о строении ДЭС на границе раздела фаз ДЭС – двойной электрический слой – тонкий поверхностный слой из пространственно разделенных электрических зарядов противоположного знака, образующихся на границе двух фаз. Если погрузить металлическую пластину (электрод) в раствор соли данного металла, то может произойти один из двух процессов. 1. Пусть металл является активным восстановителем, т. е. окисляется, тогда из-за диполей воды, содержащихся в данном растворе, какая-то часть атомов металла оставляет свои электроны на электроде и из-за процесса гидратации в виде гидратированных ионов переходит в раствор по реакции. Общий вид реакции 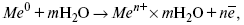 или, без учета гидратации ионов, 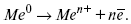 Этот процесс называется окислением. В результате него металлическая пластинка заряжается отрицательно, а катионы металла притягиваются к ней, и прилегающий к пластинке слой раствора заряжается положительно. На границе металл-раствор возникает ДЭС. Химически активные металлы как: Na, Mg, Al, Zn и др. имеют большие концентрации поверхностного раствора, состоящего из гидратированных катионов металла. При погружении в раствор своей соли любой концентрации происходит дополнительное растворение металла с образованием ДЭС, так как всегда концентрация поверхностного раствора всегда больше концентрации соли металла, и металл заряжается отрицательно. 2. Пусть металл является слабым восстановителем, тогда его ионы, содержащиеся в растворе соли, являются сильными окислителями. Некоторая часть ионов подходит к металлической поверхности и восстанавливается за счет свободных электронов, присутствующих в ней по реакции: 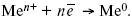 В результате процесса восстановления металлическая пластинка заряжается положительно и притягивает отрицательно заряженные частицы. При погружении малоактивных благородных металлов (Cu, Ag, Au и др.) в раствор соли наблюдается обратная картина, от первой: при любой достижимой концентрации, концентрация поверхностного раствора меньше концентрации соли металла, поэтому ионы металлов осаждаются на электроде, а в приэлектродном пространстве накапливается избыточный отрицательный заряд за счет анионов соли или ионов гидроксидной группы OH–. После достижения равновесной разницы потенциалов между металлом и раствором переход ионов в раствор прекращается. Модельные представления о строении ДЭС на границе раздела фаз: 1) модель Гельмгольца; 2) модель Гуи, Чапмена; 3) модель Штерна; 4) модель Грэма; 5) модель современная. Первая модель ДЭС открыта Гельмгольцем, он представлял ДЭС в виде двух обкладок плоского конденсатора, одна обкладка расположена непосредственно на поверхности электрода, вторая – в электролите. d = диаметру молекул Н2О. Так как все заряды сконцентрированы в двух плоскостях, то изменение потенциала по мере удаления от поверхности электрода Е0 будет описываться прямой линией. Еа – величина электродного удаления от поверхности электрода потенциала. Используя теорию конденсатора, Гельмгольц рассчитал величину заряда ДЭС и величину дифференциальной емкости ДЭС. q – величина заряда = (D/4?2)Е0, С – дифференциальная емкость ДЭС = D/4?2, D – диэлектрическая проницаемость раствора, d – расстояние между обкладками конденсатора. Теория Гельмгольца позволила объяснить ход электрокапиллярной кривой, рассчитать величину дифференциальной емкости ДЭС, хорошо совпадающую с экспериментально полученными данными. Наилучшая сходимость была получена для концентрированных растворов элементов, однако данная теория не объясняла зависимость плотности заряда и дифференциальной емкости ДЭС от состава электролита и концентрации компонента. Недостатки теории Гельмгольца: 1) не учитывалось тепловое движение ионов в растворе; 2) не учитывался размер ионов; 3) не рассматривались процессы адсорбции на границе раздела фаз (электрод – электролит). Ее применяют только к концентрированным растворам, не содержащим поверхностно-активные вещества (ПАВ). Б. Гуи, Д. Чапмен учли тепловое движение в растворах электролитов. Эта теория позволяет рассчитать плотность ?заряда ДЭС и величину дифференциальной емкости С, они учли влияние концентрации, но рассчитанные значения дифференциальной емкости С, но они меньше сходятся с экспериментально полученными результатами. Теория Гуи, Чапмена применима к разбавленным растворам электролитов. Недостатки: 1) не учитываются размеры ионов; 2) не учитывается явление адсорбции на границе раздела фаз. В. Штерн учел, что в электролитах наблюдается электростатическое взаимодействие между ионами, тепловое движение компонентов электролита и возможное специфическое взаимодействие компонентов электролита с поверхностью электрода. Он соединил теорию Гельмгольца с теорией Гуи, Чапмен, в результате ДЭС представлялся состоящим из двух частей: 1) плотной части Гельмгольца; 2) диффузной части по модели Гуи, Чапмена. За счет адсорбции ПА компонента может происходить перезаряд поверхности. Штерн считал, что адсорбция происходит на границе плотной и диффузной части ДЭС. Эта граница называется плоскостью Гельмгольца. Теория Штерна легла в основу современных представлений и развивалась в работах Грема, Фрумкина, Эршлера, Есина и др. Недостатки: 1) не учитывал дискретность зарядов; 2) величина емкости, рассчитанная по модельным представлениям Штерна, не соответствовала экспериментально полученным результатам. Г. Греем рассмотрел возможность адсорбции ПА анионов внутри плотной части ДЭС, он ввел понятие внутренней и внешней плоскости Гельмгольца. Адсорбция анионов происходит на внутренней плоскости Гельмгольца. Недостаток: рассматривал адсорбцию только анионов и не учитывал дискретность зарядов. О. А. Есин рассмотрел дискретность зарядов и показал, что ионы, образующие внутреннюю и внешнюю плоскости Гельмгольца, взаимодействуют между собой, образуя диполи. Указанное взаимодействие влияет на величину диффузной емкости с ДЭС. О. А. Есин рассмотрел возможность адсорбции на внутренней плоскости Гельмгольца как катионов, так и анионов. 8. Проводники первого и второго рода Проводники – вещества, проводящие электрический ток благодаря наличию в них большого количества зарядов, способных свободно перемещаться (в отличие от изоляторов). Они бывают I (первого) и II (второго) рода. Электропроводность проводников I рода не сопровождается химическими процессами, она обусловлена электронами. К проводникам I рода относятся: чистые металлы, т. е. металлы без примесей, сплавы, некоторые соли, оксиды и ряд органических веществ. На электродах, выполненных из проводников I рода, происходит процесс переноса катиона металла в раствор или из раствора на поверхность металла. К проводникам II рода относятся электролиты. В них прохождение тока связано с химическими процессами и обусловлено движением положительных и отрицательных ионов. Электроды первого рода. В случае металлических электродов первого рода такими ионами будут катионы металла, а в случае металлоидных электродов первого рода – анионы металлоида. Серебряный электрод первого рода Ag+/Ag. Ему отвечает реакция Ag+ + e- = Ag и электродный потенциал EAg+ /Ag = Ag+ / Ag+b0lg a Ag+. После подстановки численных значений Е 0 и b0 при 25 oС: 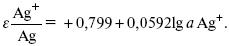 Примером металлоидных электродов первого рода может служить селеновый электрод Se2–/Se, Se + 2e- = Se2; при 25 oС ESe2–/Se0 = –0,92 – 0,03lg a Se2–. Электроды второго рода – полуэлементы, состоящие из металла, покрытого слоем труднорастворимого соединения (соли, оксида или гидроксида) и погруженного в раствор, содержащий тот же анион, что и труднорастворимое соединение электродного металла. Схематически электрод второго рода можно представить так: АZ–/MA, M, а протекающую в нем реакцию – МА + ze = М + АZ–. Отсюда уравнением для электродного потенциала будет: 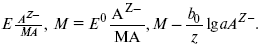 Каломельные электроды – это ртуть, покрытая пастой из каломели, и ртуть, находящаяся в контакте с раствором KCl. Cl– / Hg2Cl2, Hg. Электродная реакция сводится к восстановлению каломели до металлической ртути и аниона хлора: 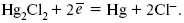 Потенциал каломельного электрода обратим по отношению к ионам хлора и определяется их активностью: 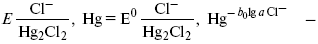 При 25 оС потенциал каломельного электрода находят по уравнению: 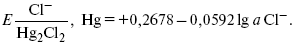 Ртутно-сульфатные электроды SO42 –/Hg2SO4, Hg аналогичны каломельным с той лишь разницей, что ртуть здесь покрыта слоем пасты из Hg и закисного сульфата ртути, а в качестве раствора используется H2SO4. Потенциал ртутно-сульфатного электрода при 25 oС выражается уравнением: 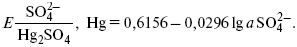 Хлорсеребряный электрод представляет собой систему Cl–/AgCl, Ag, а его потенциалу отвечает уравнение: ECl– /AgCl, Ag = E0Cl–/AgCl, Ag –b lg aCl– или при 25 оС: ECl–/AgCl, Ag = 0,2224 – 0,0592 lg a Cl–. 9. Электроды сравнения Электроды сравнения – электроды, используемые при измерении электродных потенциалов в паре с используемым электродом. Электродный потенциал – скачок потенциала на границе металл-раствор. Он определяется: природой металла, раствора, концентрацией, температурой. Для сравнения электродных потенциалов нужны стандартные условия: t = 25 °С = 298 К; Р – 1 атм, одномолярный раствор. Абсолютное значение электродного потенциала измерить нельзя. Поэтому измеряют разность потенциалов между данным электродом и электродом сравнения, потенциал которого принимают равным нулю. Часто используют водородный электрод, изготовленный из губчатой платины с сильно развитой поверхностью (платиновая чернь), опускают в раствор H2SO4 – серной кислоты с активностью ионов водорода, равной единице. При этом через раствор пропускается газообразный водород под давлением, который затем адсорбируется платиной. Относительно потенциала водородного электрода все металлы располагают в ряд напряжений, установленный электрически Н. Н. Бекетовым, взаимному вытеснению металлов в зависимости от величины и знака стандартного электродного потенциала. Существуют и другие электроды сравнения: каломельный, хлорсеребряный и другие, в зависимости от различных методов. Конструктивное оформление электрода сравнения разнообразно. Например, для полярографического метода электроды сравнения должны иметь большую поверхность во избежание поляризации их при работе под током. |
|
||
|
Главная | В избранное | Наш E-MAIL | Добавить материал | Нашёл ошибку | Вверх |
||||
|
|
||||